Running FOLD-EM
Here we show an example of building a backbone model, using FOLD-EM, for a 6.8 Å Rice Dwarf Virus (RDV) cryoEM map from [Z+01]. Follow the same steps for any other density map.
Software Download Site
Email us for the current FOLD-EM software source code snapshot.
It is now combined with the MOTIF-EM package. So you should get the same stuff from the MOTIF-EM site and hence the installation instructions are same as for MOTIF-EM: http://ai.stanford.edu/~mitul/motifEM/compiling.html
Limitations and scope of the FOLD-EM software:
Estimated Run Time:
FOLD-EM took about 4 days on a computing cluster comprising of 100 nodes for this testcase.
Getting started to run FOLD-EM:
1. Prepare “Domain Database”
FOLD-EM compares an input cryo-EM map with an existing database of protein domains, to construct a backbone model for the input map. We will refer to this database as DD from now on.
Now create an input file “list.inp” which contains the list of domains (or entries in the DD) you want to build the backbone from. An example of “list.inp” is here. The following list of lines is another example:
1kp8 A 2 136 A 410 526
1kid A -1 -1
1kp8 A 137 190 A 367 409
1dlw A -1 -1
1kr7 A -1 -1
3sdh A -1 -1
1phn A -1 -1
1nek B 107 238
1gte A 2 183
1grj A 2 79
The first column is the pdb name. The second is the chain id and the following two columns corresponds to start and end amino acid numbers, and so. As seen also, some domains can have multiple segments, like in the case of 1kp8, in line #3.
For the experiments in our paper [SM11], we used the SCOP database to build a DD that approximately represent all the domains in SCOP. DD comprising of about 4000 representative protein domains from the SCOP database, was build as follows. The first member of a domain family is picked as the representative structure, and additional structures from the same domain family are included if they structurally differ by > 5 Å RMSD from each other. These structures represent all super families of the five SCOP-domain classes: all-alpha, all-beta, alpha+beta, alpha and beta, and small proteins. The “list.inp” corresponding to this DD is here.
Once you have “list.inp”, you can download the actual domains from SCOP using the following command, unless you have already done so:
$LSC/download.n.break.nohets.pl list.inp $DATABASEDIR
-- $FOLDEM is the directory where the FOLD-EM package was unpacked.
-- $MOTIFEM is $FOLDEM/pattern_matching
-- $LSC is $MOTIFEM/large_scale_matching
-- $DATABASEDIR is the directory where you want to store the database
(If you like, you can set the variables using “setenv” or “export”. BUT PLEASE AT-LEAST SET $FOLDEM)
Now the next step is to do some database pre-processing, which will be used in any FOLD-EM run that uses DD. So, securely store the pre-processing results for future uses of FOLD-EM.
2. Database Pre-processing
- Since this step is independent of the input map, you may not need to do this step if you saved the output of this step from a previous fold-em run.
- You will need to install EMAN [LBC99], as we will use the tool “pdb2mrc” from it.
- Go to $DATABASEDIR and run:
$LSC/pdb2xplor.all.pl list.inp $DATABASEDIR <resolution in integral value> 1
- The parameter <resolution in integral value> resolution takes values from 5 to 20. That is, you run the above command for each parameter value. It can take a a while to finish running all. You can look at the output being generated while running to see how much of the execution is left. A given run goes through each line of “list.inp” one by one. So by looking at the outputs you can figure out, on which line of “list.inp” is the execution on, at a given moment.
- BUT, you can do this pre-processing only partially now, leaving rest for the future, as it takes a while to finish them all. For instance, you can just run the above command for the <resolution in integral value> value of 7, if you just want to test the up-coming testcase of building a backbone model for RDV. That is, if you just want to run FOLD-EM on a cryo-EM map with resolution X (Angstrom) for now, then you can do the above run for <resolution in integral value> value of X for now, and do rest of the runs latter. You can save this partially pre-processed database for future FOLD-EM runs.
-At this point each run of above command, for a given resolution, generates about XXXXX bytes of data.
Run the second pre-processing step:
$LSC/run_full.gen_kps_grads.2.pl list.inp $DATABASEDIR <resolution in integral value> 1
- Make sure $DATABASEDIR is full path and not relative path. Also make sure the environment variable $FOLDEM is set as mentioned above.
- <resolution in integral value> is the resolution values for which you have run the previous step.
This would generate a file of jobs “jobs.txt.-1.-1”.
- Then on a computing cluster run the jobs in “jobs.txt.-1.-1” using run_coms.MPI:
mpiexec -n <numprocs> $MOTIFEM/MPI_implementation/run_coms.MPI jobs.txt.-1.-1
-- where <numprocs> is the number of processors you want use (at-least 2 and at-most (number domains in list.inp + 1))
-- here is a typical job file that I use to run this in my cluster (As seen in this job file, always make sure paths are set and libraries linked, correctly, as per http://ai.stanford.edu/~mitul/motifEM/compiling.html, before running motif-em/fold-em; You may need to ask for different number of processors than what is asked in this job file).
- BUT, you can do this pre-processing only partially now, leaving rest for the future, as it takes a while to finish them all. For instance, you can just run the above command for the <resolution in integral value> value of 7, if you just want to test the up-coming testcase of building a backbone model for RDV. That is, if you just want to run FOLD-EM on a cryo-EM map with resolution X (Angstrom) for now, then you can do the above run for <resolution in integral value> value of X for now, and do rest of the runs latter. You can save this partially pre-processed database for future FOLD-EM runs.
-At this point each run of above command, for a given resolution, generates about XXXXX bytes of data.
- If it’s successful, then for every …
3. Now running FOLD-EM on a test-case
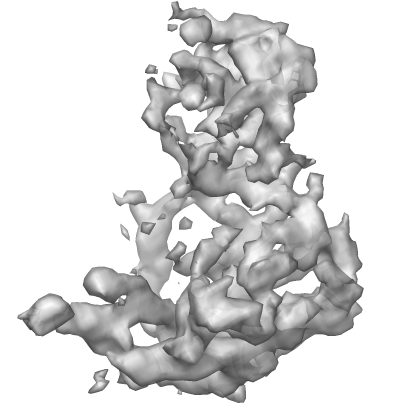
Now let’s do a test run on FOLD-EM. We will use FOLD-EM to build a backbone for a 6.8 Å RDV map from [Z+01]. You can download the map from here.
This is how the RDV map looks like in Chimera:
- Create a work directory $WORKDIR, and go into it (do “cd”) to run the following commands .
- FOLD-EM takes input cryo-EM maps in the XPLOR format. Download the RDV XPLOR map from here. Let’s call it “inp.xplor” here.
- Remember the resolution of the input map, which is 7 Å for the RDV input map here. This resolution will be the input parameter for some of the commands to follow.
-Now pre-process the input map:
-- first run the following command
$LSC/preprocess_input_map.3.pl $WORKDIR inp.xplor $MOTIFEM
-- make sure inp.xplor is in $WORKDIR
-- Make sure $WORKDIR is full path and not relative path
-- As mentioned earlier, $MOTIFEM should be “$FOLDEM/pattern_matching”, where $FOLDEM is the directory where the FOLD-EM package was unpacked.
-- Then on a computing cluster run:
mpiexec -n <numprocs> $MOTIFEM/MPI_implementation/motifem.svds.kps 0 1
--- where <numprocs> is the number of processors you want use
--- make sure the working directory for this step is “$WORKDIR/input_map_processing/” (after this step you return to $WORKDIR)
--- here is a typical job file that I use
- Now do a set-up:
$LSC/set_up.pl list.inp $DATABASEDIR $WORKDIR inp.xplor 7 1
- Make sure $WORKDIR and $DATABASEDIR are full paths and not relative paths
- the parameter 7 is the resolution of the input RDV map used here. Change accordingly.
- If it’s successful, then for every …
- it takes about XXXX minutes
-Generate job files:
$LSC/run_full.motifem.1shot.3.pl list.inp $WORKDIR
- This would generate a file with jobs “jobs.m1s.txt.-1.-1”. Then on a computing cluster run the jobs in “jobs.m1s.txt.-1.-1” using run_coms.MPI:
mpiexec -n <numprocs> $MOTIFEM/MPI_implementation/run_coms.MPI jobs.m1s.txt.-1.-1
-- where <numprocs> is the number of processors you want use (at-least 2 and at-most (number domains in list.inp + 1))
-- here is a typical job file that I use to run this in my cluster (As seen in this job file, always make sure paths are set and libraries linked, correctly, as per http://ai.stanford.edu/~mitul/motifEM/compiling.html, before running motif-em/fold-em; You may need to ask for different number of processsors that what is asked in this job file). If you need to include time limit in your job file, then a typical run of a job in “jobs.m1s.txt.-1.-1” on this RDV map can take 2-6 hours.
-If all runs are successful, then should see a file named “allscores.txt” in all the sub-directories of $WORKDIR
-Now analyze results :-
$LSC/run_full.PBS.analyze.pl list.inp $WORKDIR 7 1
- the parameter 7 is the resolution of the input RDV map used here. Change accordingly.
This will give the file “results.txt”, containing the list of candidate domains
Now, let’s select some domains from “results.txt”, that fit the RDV map “inp.xplor.
-- Open it and pick up the domain you like, as the first domain. Most probably you will like the first domain listed there, or maybe one in top 10. (The score at the last column is the XXXX score ...)
-- You can look at “results.txt” and manually pick up more domains as it suits you. But you can also autmatically choose subsequent domains using the command:
select_domain_number_i_from_results.txt <map.xplor> <map.xplor threshold> <file(containing pdb names), e.g. “results.txt”> <window; typically 4A> <overlap threshold; typically: 0.5> <which domain: N:1,2,3..> <number of solutions> <list of N-1 domains>
-- <map.xplor threshold> is the map threshold value next to parameter “zero_thresh_o”, in the file “$WORKDIR/input_map_processing/inp.txt”
-- <which domain: N:1,2,3> is the Nth domain you want to choose. <list of N-1 domains> is the list of (N-1) domains you have already chosen. The list is domains picked from the first column of “results.txt”
e.g., suppose for the RDV, if you choose the first domain from “results.txt” as the first domain, then you can choose the 2nd domain automatically by:
select_domain_number_i_from_results.txt inp.xplor 1.19 results.txt 4 0.5 2 5 <entry in (row 1, column 1) of “results.txt”>
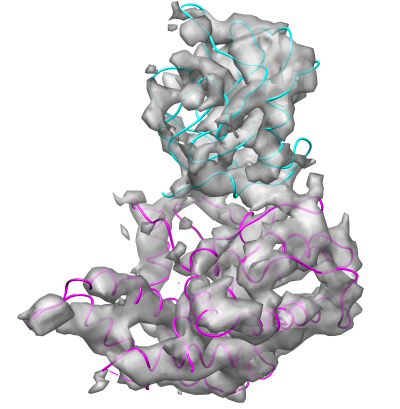
This is how the final fitting, obtained by FOLD-EM, of the two discovered domains (the two coloured ribbon structures) looks like in Chimera:
References
[Z+01] Zhou, Z.H. et al. Electron cryomicroscopy and bioinformatics suggest protein fold models for rice dwarf virus. Nat Struct Biol 8, 868-873 (2001).
[LBC99] Ludtke, S.J., Baldwin, P.R., and Chiu, W. (1999). EMAN: semi-automated software for high-resolution single-particle reconstructions. J Struct Biol 128, 82-97.