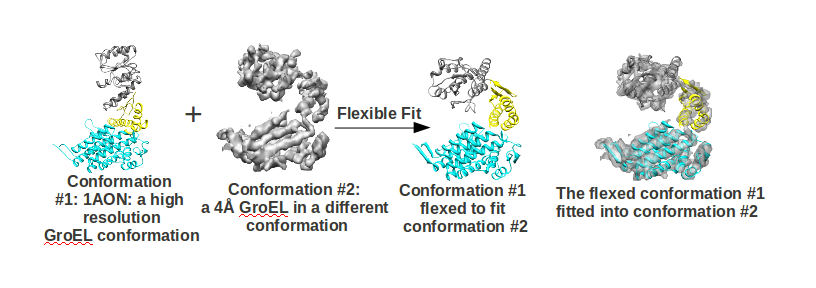
Automated Flexible Docking/Fitting of
Structures (Atomic Resolution or Low Resolution) into Cryo-EM Maps
Scope and Limitation (DO HEED THIS)
- Same as for MOTIF-EM: http://ai.stanford.edu/~mitul/motifEM/compiling.html
- The domain extractions are not perfect. Hence some residues might be missing from the extracted domains. Also extracted domains would be disconnected. That is, the software does NOT connect the domains with linkers.
Preperation
At this point it is assumed that you have installed MOTIF-EM as per: http://ai.stanford.edu/~mitul/motifEM/compiling.html
Additionally, you will need to install EMAN [LBC99], as we will use the tool “pdb2mrc” from it.
Copy the files “utility_codes/flex_fit_job_file_template.csh” and “utility_codes/flex_fit_job_file_template_submission_command.txt” from the MOTIF-EM installation directory into the current work directory. Modify “flex_fit_job_file_template.csh” as per your computing cluster’s specifications. Make sure the number of computing CPUs you specify is the same number you specify latter while running the commands. “flex_fit_job_file_template_submission_command.txt” contains the command to submit this job file in a computing cluster. Modify as per your computing cluster.
Running Flexible Fitting
Then simply run:-
flex_fit.v4.pl <conformation1.pdb> <conformation2.xplor> <resolution of conformation2.xplor> <numprocs> <number of rounds>
- <conformation1.pdb> is the atomic resolution conformation you want to flex fit
- <conformation2.pdb> is the low resolution structure in XPLOR format into which you want to flex fit <conformation1.pdb>
- <resolution of conformation2.xplor> is the resolution of <conformation2.xplor> in Å
- <numprocs> is the number of processors available to be used. Make sure this is same as you what you specify in “flex_fit_job_file_template.csh”
- <number of rounds> is the number of domains it will search for.
The final generated fits will be of format: “sol*trans.pdb”
References
[SM12] Saha, M, Morais MC. FOLD-EM: automated fold recognition in low resolution electron density maps. To be published.
[SLC10] Saha, M., Chiu, W. & Levitt, M. MOTIF-EM: an automated computational tool for identifying conserved regions in CryoEM structures. M. Bioinformatics 26 (12), 301-309 (2004).
http://cs.stanford.edu/~mitul/motifEM/
[LBC99] Ludtke, S.J., Baldwin, P.R., and Chiu, W. (1999). EMAN: semi-automated software for high-resolution single-particle reconstructions. J Struct Biol 128, 82-97.