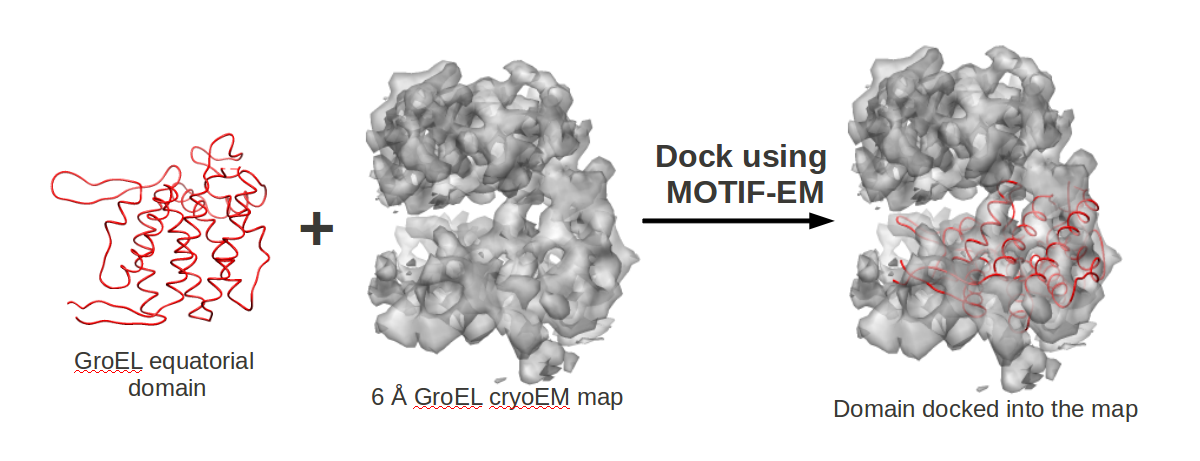
Docking/Fitting a Domain (Atomic Resolution or Low Resolution) into a Cryo-EM Map using MOTIF-EM
Here we show how to use MOTIF-EM http://ai.stanford.edu/~mitul/motifEM/ to do docking/fitting of atomic resolution structures/domains (or even low resolution cryoEM structures) into low resolution structures from Cryo-EM or electron density maps.
Advantages of using MOTIF-EM to dock/fit compared to other software (as of 2011)
Scope and Limitation (DO HEED THIS)
Same as for MOTIF-EM: http://ai.stanford.edu/~mitul/motifEM/compiling.html.
Preperation
At this point it is assumed that you have installed MOTIF-EM as per: http://ai.stanford.edu/~mitul/motifEM/compiling.html
Additionally, you will need to install EMAN [LBC99], as we will use the tool “pdb2mrc” from it.
Generate the input file “inp.txt” by running this command:
set_up_domain_fit.pl <map.xplor> <domain.pdb> <map_resolution> <work directory> <MOTIF-EM source directory>
- <map.xplor> is the input cryoEM map in XPLOR format
- <domain.pdb> is the input high resolution PDB file of a domain you want to fit in the map
- <map_resolution> is the resolution of the cryoEM map in Å
- <work directory> is the directory you want to work in
- <MOTIF-EM source directory> is the MOTIF-EM installation with full path
If the domain you want to fit is another cryo-EM map, then instead use this command:
set_up_map_fit.pl <map.xplor> <domain.xplor> <work directory> <MOTIF-EM source directory>
- <map.xplor> is the input cryoEM map in XPLOR format
- <domain.xplor> is the input low resolution domain you want to fit in the map
- <work directory> is the directory you want to work in
- <MOTIF-EM source directory> is the MOTIF-EM installation with full path
Running MOTIF-EM
Now, in the current working directory where “inp.txt” is located, run MOTIF-EM (the Step -4- of the MOTIF-EM running steps at: http://ai.stanford.edu/~mitul/motifEM/run_commands.html)
Analyzing the results
Now many fits are generated. To see the first N fits, run:
generate_N_fits.pl <domain.pdb> N
- <domain.pdb> is the input high resolution PDB file of a domain
The fits are generated as “<domain.pdb>_fit_1.pdb”, “<domain.pdb>_fit_2.pdb”, and so on.
Now, only a part of domain might have fitted onto the map. So <domain.pdb>_fit_i.pdb.part.pdb is that part of <domain.pdb>_fit_i.pdb. This shows the partial matching/fitting strength of MOTIF-EM.
However, to generate only the i-th fit, run:
generate_fit.pl i <domain.pdb> <domain.fitted.pdb>
- <domain.pdb> is the input high resolution PDB file of a domain
- <domain.fitted.pdb> will be domain.pdb fitted onto the input map as per the i-th solution.
If the domain you are fitting is another cryo-EM map, then instead use this command:
To see the first N fits, run:
generate_N_map_fits.pl <domain.xplor> N
- <domain.xplor> is the input low resolution domain you want to fit
The fits are generated as “domain.xplor_fit_1.xplor”, “domain.xplor_fit_2.xplor”, and so on.
Now, only a part of domain might have fitted onto the map. So <domain.xplor>_fit_i.xplor.part.xplor is that part of <domain.pdb>_fit_i.xplor. This shows the partial matching/fitting strength of MOTIF-EM.
However, to generate only the i-th fit, run:
generate_map_fit.pl i <domain.xplor> <domain.fitted.xplor>
- <domain.xplor> is the input low resolution domain you want to fit
- <domain.fitted.xplor> will be domain.xplor fitted onto the input map as per the i-th solution.
References
[SLC10] Saha, M., Chiu, W. & Levitt, M. MOTIF-EM: an automated computational tool for identifying conserved regions in CryoEM structures. M. Bioinformatics 26 (12), 301-309 (2004).
http://cs.stanford.edu/~mitul/motifEM/
[LBC99] Ludtke, S.J., Baldwin, P.R., and Chiu, W. (1999). EMAN: semi-automated software for high-resolution single-particle reconstructions. J Struct Biol 128, 82-97.