in Electron Density Maps
After a sufficiently high-resolution cryoEM reconstruction of a macromolecular assembly has been determined, the next crucial step is to build backbone models of its macromolecular components. We present the first fully automated computational tool, FOLD-EM, to build backbone models for newly generated cryoEM structures by identifying component domains (by refering to an input domain databank, such as SCOP) in electron density maps that can then be combined into larger protein folds and higher-order structures. FOLD-EM, however, does not connect the identified domain components to yield a singly-connected backbone chain.
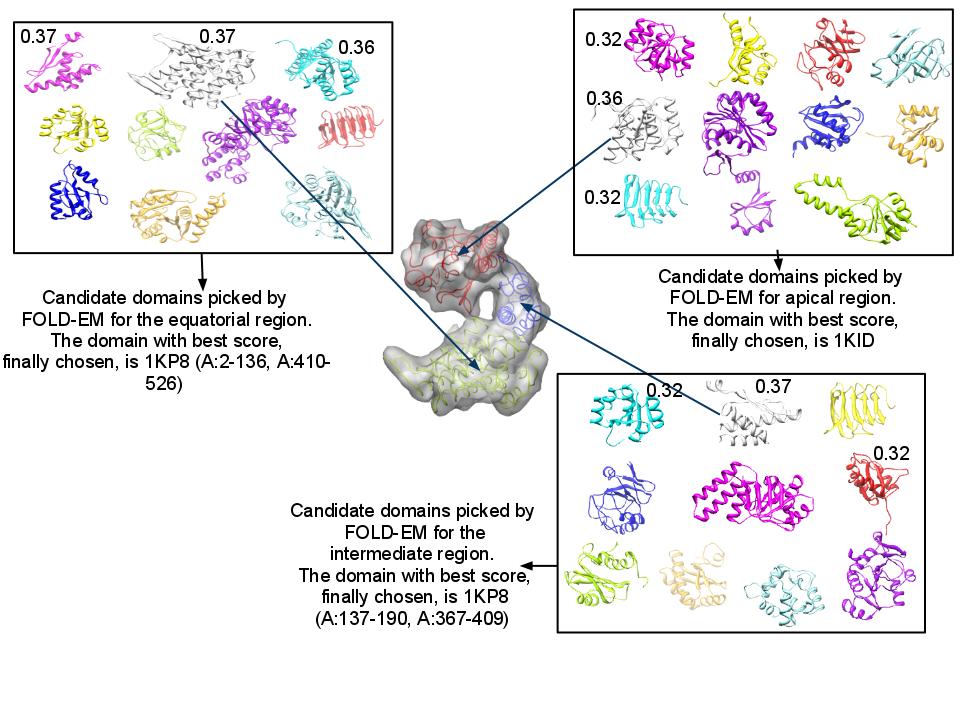
- [Current FOLD-EM Software Snapshot and a Fold Recognition Testcase]
- [Docking/Fitting of Atomic Resolution Structures into Low Resolution Structures/Maps]
- [Flexible Docking/Fitting of Atomic Resolution Structures into Low Resolution Structures/Maps]
- [Early paper version]
- [Official Publication: "FOLD-EM: automated fold recognition in medium- and low-resolution (4–15 Å) electron density maps", Bioinformatics (2012) 28(24):3265-3273. Keywords: cryo-EM maps, structure comparison, partial matching, SIFT, domain docking, flexible fitting, large scale comparison, SCOP, backbone modeling]